课件共330页,为《量子化学计算入门》课程内容,为了帮助理论计算初学者及对计算感兴趣的实验人员快速建立学习构架,免费分享给大家。
内容完美契合零基础小白,带你横扫理论盲区,讲解4大计算模拟理论、10类电子结构计算、21款常用软件、VASP计算入门案例。
通过理论联系实际,带大家了解各类软件特点与适用范围,选择适合自己的工具与计算方法,快速入门DFT计算。以下为部分课件展示!
添加下方微信好友回复“330”,免费下载!
课件前两部分侧重理论,涉及分子轨道理论、密度泛函、分子动力学理论等4大计算材料学理论,单点能、态密度、能带计算等10类电子结构性质计算。后两部分侧重应用,详细介绍了VASP、Materials Studio、Gaussian 16、Quantum ESPRESSO等21种分子模拟软件的应用、优缺点及适用范围。然后,针对VASP软件做了计算流程、输入文件、输出文件讲解,最后以表面吸附为例,给出结构优化、吸附能、态密度及能带计算方法与注意事项。1、从分子轨道理论到Hartree-Fock理论
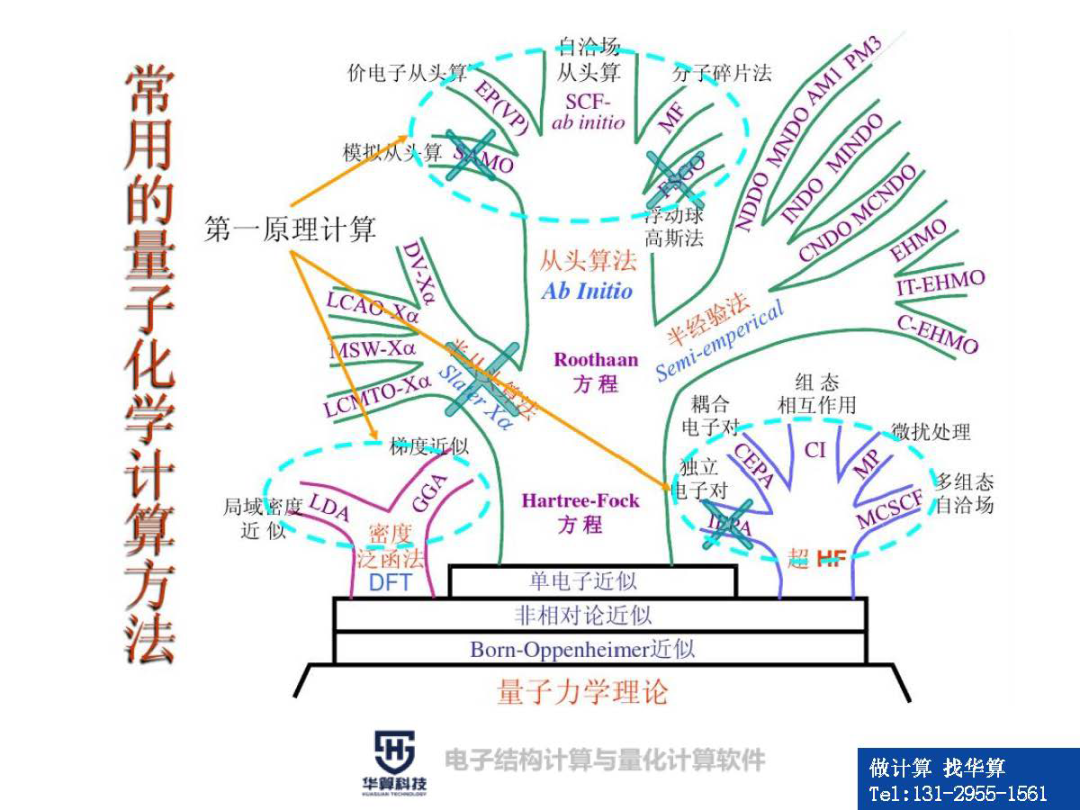
(1)Born-Oppenheimer(BO)近似(5)Hartree-Fock-Roothaan方程(6)两种典型基函数:斯莱特基(STO)与高斯基(GTO)2、后Hartree-Fock(HF)电子相关方法
3、密度泛函理论
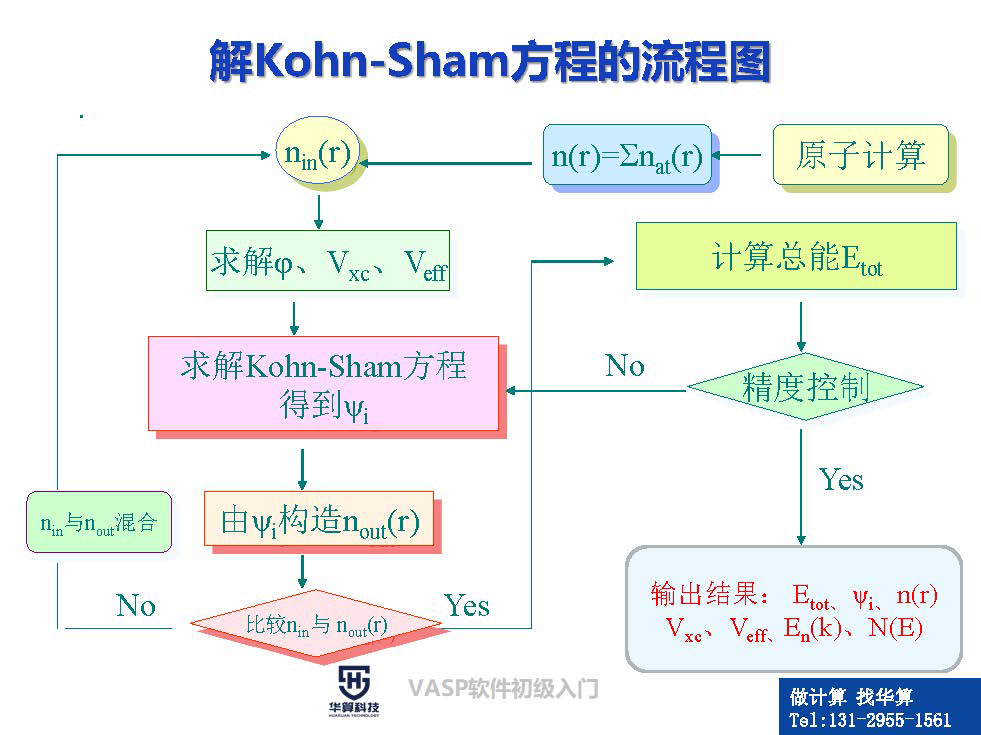
(2)Hohenberg-Kohn定理及Kohn-Sham方程及其求解(10)DFT中其它典型误差:自相互作用修正(SIC);离域化误 差;静态关联误差 4、分子动力学理论
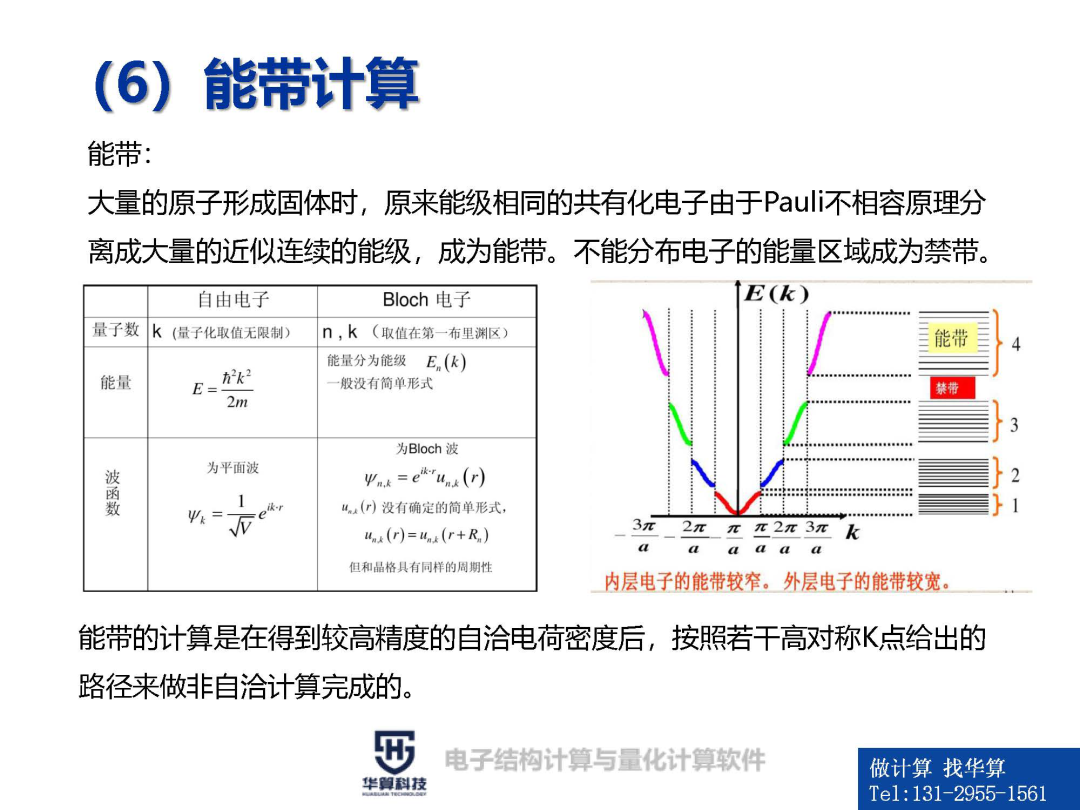
(2)分子力场:详细介绍传统力场、第二代力场、通用力场、极化力场及粗粒化 力场;分子力场的选择;力场存在问题;力场的发展趋势1、电子结构计算
2、常用量子化学软件
涉及8款量子化学软件的分类、应用及优缺点介绍,具体包括: Gaussian 16,MOLPRO,MOLCAS,TURBOMOLE,Q-chem,NWchem,MOPAC,ADF, 3、其它计算材料学软件
涉及13款量子化学软件的分类、应用及优缺点介绍,具体包括:VASP,Material Studio,CASTEP,DMol3,Wien2K,ABINIT,Quantum-Espresso,SIESTA,ATK,Lammps,GROMACS,CPMD,Chemshell1、VASP计算流程
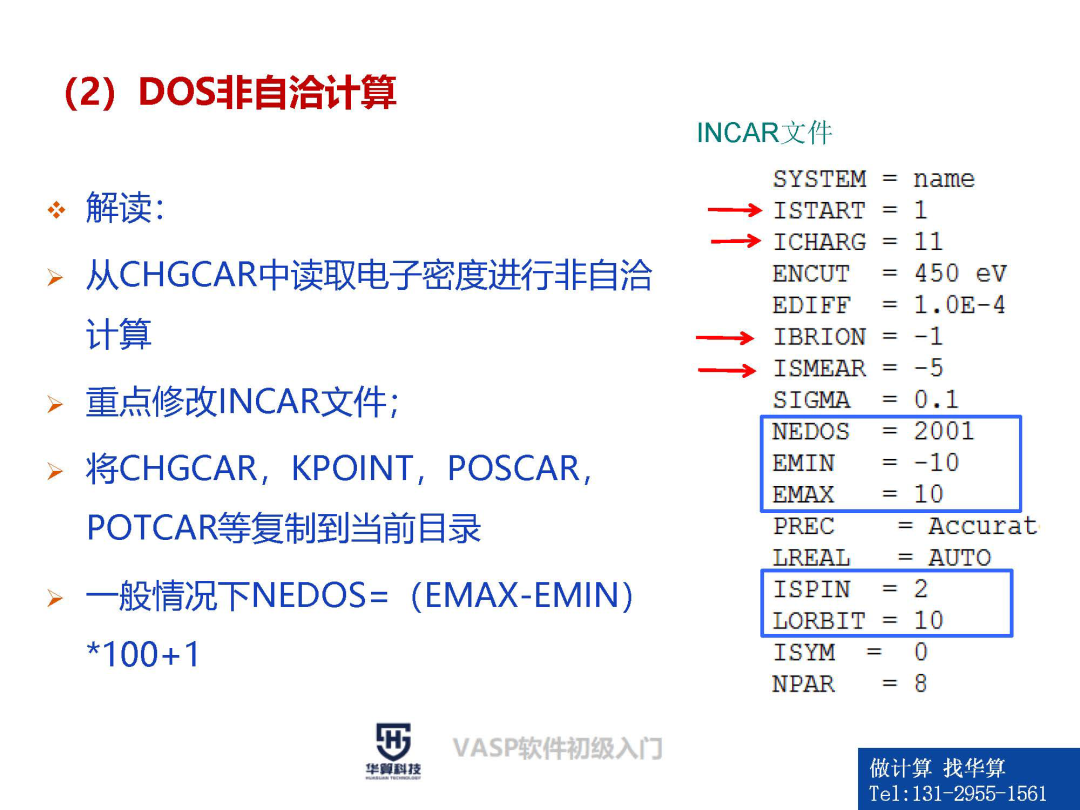
2、VASP输入文件及其重要参数详解
4、实例讲解——MoS2表面吸附Ag原子