继上期的软文分享之后,这次小编又为大家推荐一波免费的生物信息学分析软件。

细绳
蛋白质相互作用网络
String 是一个已知和预测的蛋白质相互作用数据库,该数据库可应用于 2031 个物种,包括 960 万种蛋白质和 1380 万种蛋白质间相互作用,除了实验数据、PubMed 摘要文本挖掘结果和其他数据库数据外,还包含使用生物信息学方法预测的结果。
下面简单介绍一下操作。
在主页上,您可以通过多种方式进行搜索:
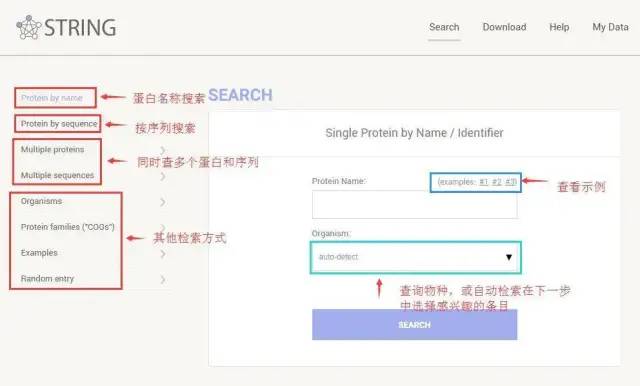
我们来举个例子,搜索之后会出现一个图,图中的每个节点代表一种蛋白质,默认情况下,节点的颜色分为红白两色,红色代表查询蛋白质,白色代表与查询蛋白质有相互作用的其他蛋白质。由于白色不太好看,string 会根据相互作用的分值来映射颜色。
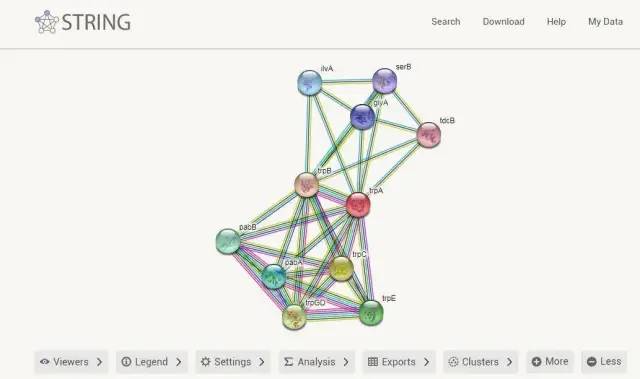
在图例页面中可以看到每个蛋白质的颜色和对应的分数值,如下图所示:

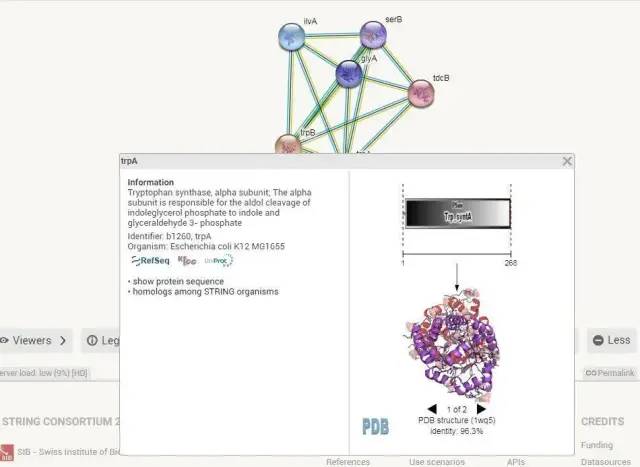
点击圆圈节点可以显示该蛋白质的具体信息,如下图:
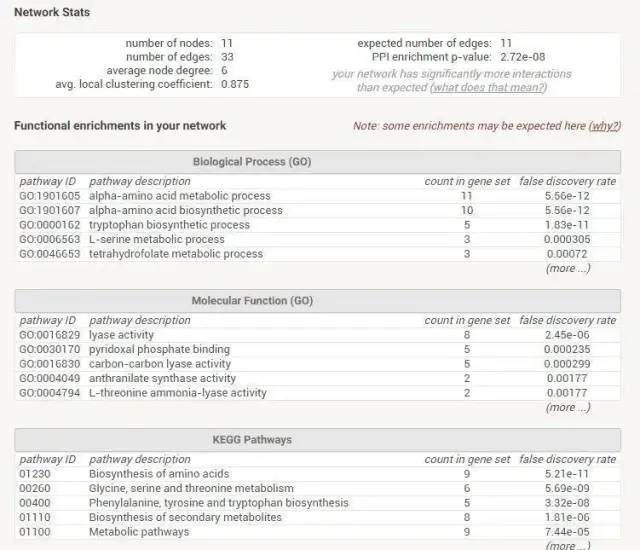
在分析页面,提供了蛋白质相互作用网络中基因的GO和KEGG富集分析结果,如图所示:

联合通讯社
基因共表达网络
Coexpedia 可以对单个或多个基因进行共表达网络分析。
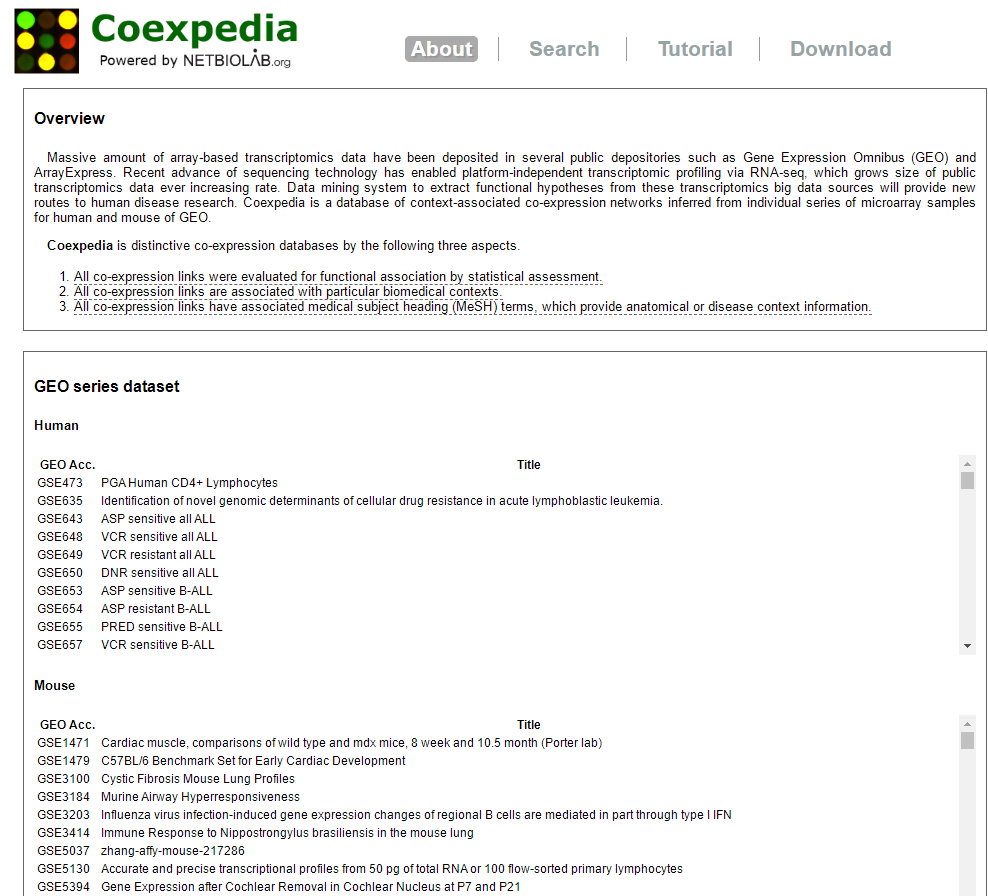
但该网站只有人类和小鼠的数据。
下面简单介绍一下操作。如果我们要制作一个CTLA4共表达网络,
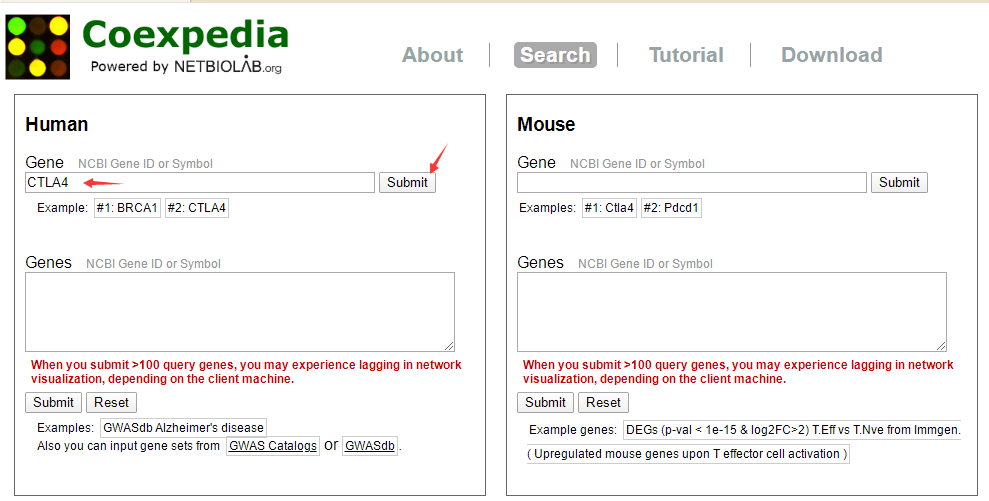

相关数据也可以下载。
模因
序列基序
MEME是一个包含多个软件的工具包,MEME是一个用于motif挖掘的软件,MEME不允许motif中存在空缺,MAST是对MEME的后续分析,可以在MEME结束后通过超链接继续进行,也可以将meme保存为文本格式的文件。
网络日志
创建序列徽标图
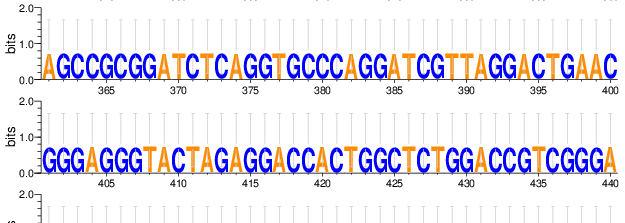
序列标识是序列的标志,是将序列比对中各个位置上的残基按顺序绘制出来的图形,各个位置上残基的积累可以反映出该位置上残基的一致性。每个残基对应的图形字符的大小与该位置上残基的频率成正比,但图形字符的大小不等于频率百分比,而是经过简单的统计计算后换算的结果。
我们可以在“序列数据输入”中输入一个序列来生成序列识别图。
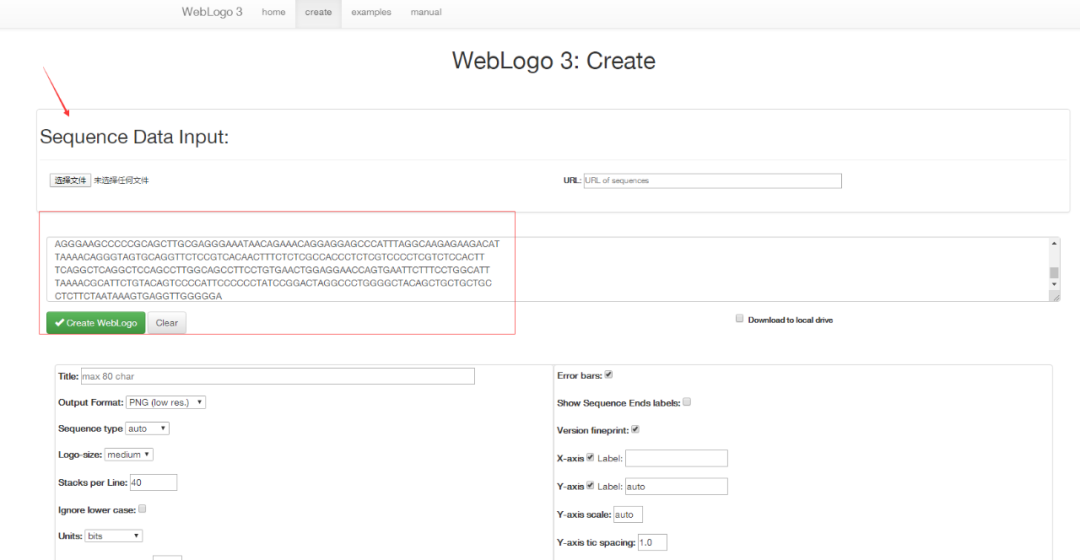
基因卡
基因信息搜索
Genecards可以检索人类基因信息,整合了大量的文献信息,涵盖了多个数据库中基因的分析数据,包括了任何与基因相关的信息。(温馨提示,使用前记得注册账号!)
ESPript
顺序显示
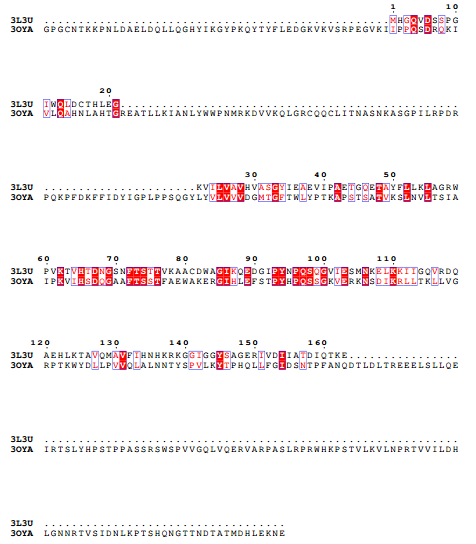
ESPript(Easy Sequencing in Postscript)是一个基于比对序列呈现相似性和二级结构信息的程序。
获取序列比对文件(可通过CLUSTALW下载)后,即可打开ESPript网站绘制序列比对结果。点击Select File上传clustalw.aln文件,然后点击页面顶部的Submit按钮,在页面底部选择输出文件类型和图片格式(PNG和TIF)。
完成后会自动弹出结果页面,本文只勾选PDF文档,所以只输出以下两个文件,任意一个都可以打开。
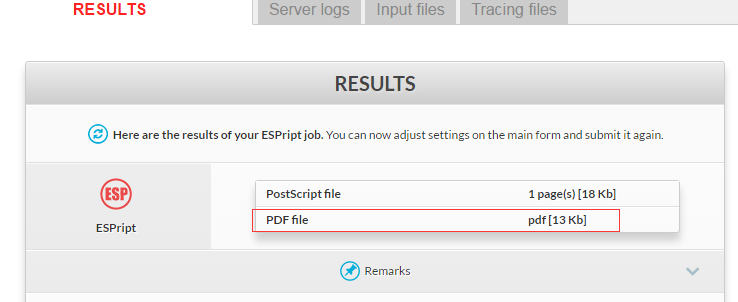
最终结果文件内容如下,可以用PS等图形处理软件进行处理和加工,比如减少两行序列之间的距离。
比奥玛特
GeneID 转换
BioMart是一个R包,可以使用Biomanager进行安装。BioMart可以找到某个基因在染色体上的位置;通过EntrezGene ID找到相关序列的GO注释;通过EntrezGene ID找到相关序列上游100bp序列(可能含有启动子等调控元件);找到人类染色体各个区域内已知的SNP;给定一组序列ID,获取其中的特定序列。
下面做简单解释。
安装 bioMart 包
install.packages("BiocManager")BiocManager::install("biomaRt")
加载 biomaRt 并显示可连接的数据库
library("biomaRt")listMarts()
##用useMart函数选定数据库mart<-useMart("emsembl")##用listDatasets()函数显示当前数据库所含的基因组注释listDatasets(mart)##用listFilters()函数查看可选择的输入类型listFilters(mart)##用listAttributes()函数选定输出的ID类型listAttributes(mart)##用getBM()函数获取注释hg_symbols<- getBM(attributes=c('ensembl_gene_id','hgnc_symbol',"chromosome_name", "start_position","end_position", "band"), filters= 'ensembl_gene_id', gene = my_ensembl_gene_id, mart = mart)
文尼2.0
维恩图
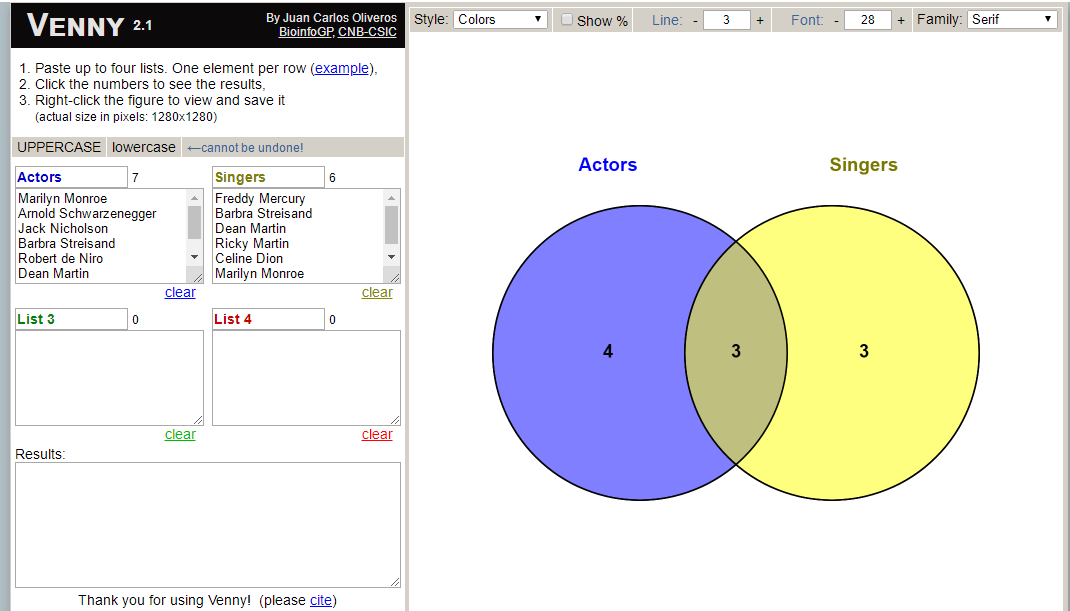
维恩图用于表示几组数据之间的关系,常见于转录组分析中差异表达基因的统计结果。这款在线工具支持最多4组数据的绘制维恩图。操作非常简单,只需要将几组数据的列表输入到对应的列表中,就可以自动生成维恩图。通常我们只需要修改列表名称,将Style改为Colors,就可以生成一张简洁美观的维恩图。图片导出也很简单,在图片框中右键,将图片另存为即可。
进化论
Evolview可以在线进行系统的进化分析,简单但不简单,可以标注比较复杂的注释信息,并导出多种格式的文件,包括pdf、svg、tiff、png,而且完成的进化树可以永久保存在网站上,也可以分享。(温馨提示,使用前记得注册账号!)
需要导入 Newick、NHX、PhyloXML 和 Nexux 等格式的纯文本文件。

工具栏包括基本设置、高级设置、添加注释信息和导出文件。
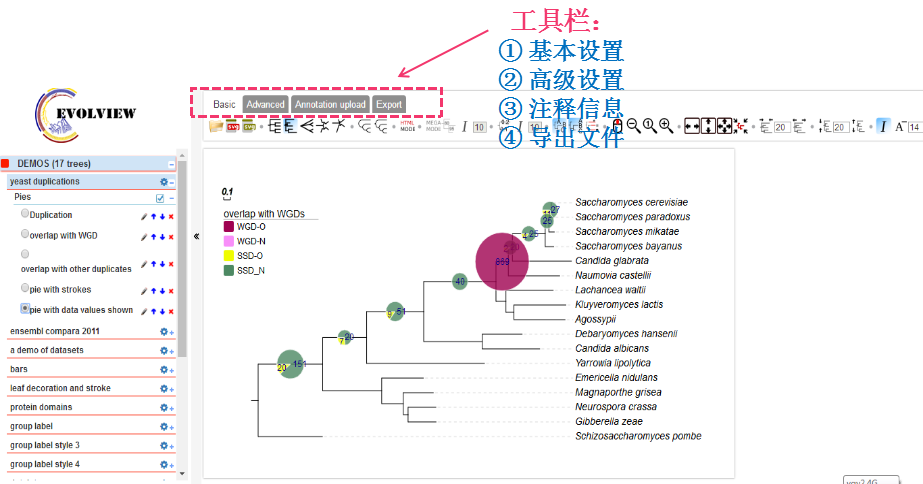
最终的效果如下:
推广者扫描
启动子预测
根据基因组中转录因子结合位点分布的不平衡性,利用转录因子结合位点分布密度与TATA box权重矩阵相结合的方法从基因组DNA中识别出启动子区域。但上述程序预测的假阳性率较高,在PROMOTER 210中每23kb出现一个假阳性,在PRO2MOTER SCAN中每19kb出现一个假阳性。
预测蛋白质
蛋白质二级结构预测
该工具可以得到很多蛋白质序列的结构信息,如功能预测、二级结构、基序、二硫键结构、结构域等。该方法的平均准确率超过72%,最佳残基预测准确率超过90%,因此被视为蛋白质二级结构预测的标准。(温馨提示,使用PredictProtein工具前,用户需先注册ID并验证邮箱。)
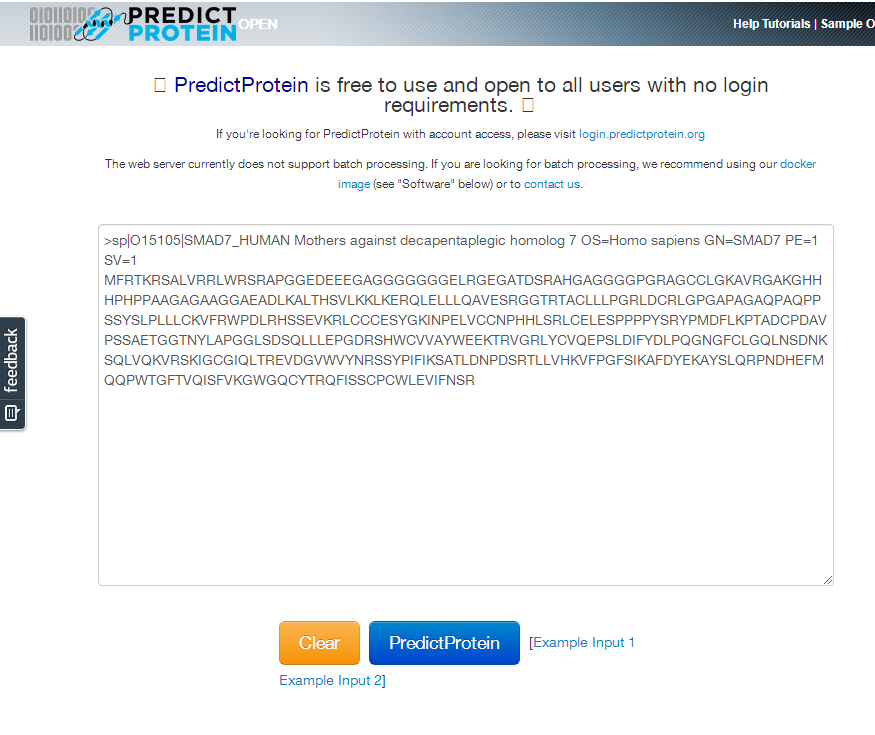
属性值
ProtParam可以预测蛋白质的基本物理化学性质。
奥米科
GO富集分析工具
基因本体(Gene Ontology,GO)是一套国际标准化的基因功能分类体系,提供一套动态更新的标准词汇,全面描述生物体内基因和基因产物的属性。GO共有三个本体,分别描述基因所涉及的分子功能、细胞成分和生物过程。GO的基本单位是条目和节点,每个术语对应一个属性。
我们可以使用Kidio OmicShare云平台来进行GO分析,做GO分析的时候需要准备两个数据文件:富集目标基因文件和GO背景基因文件。
下面简单介绍一下操作:
步骤1:上传目标基因文件;
第2步:选择(或上传)背景基因文件;
步骤3:提交。
方法一:

方法二:使用自己的背景文件进行GO富集分析
输出:
1. out.[PFC].html网页格式结果,3个对应GO的3个主要类别。结果如下图所示,包括两部分:
第一部分是GO富集结果统计表,包括GOid、GO功能描述、基因占比、背景基因占比、P值、Q值,其中P值和Q值小于0.05的以红色显示。

第二部分是GO中富集的特定基因。点击GOid可以链接到该基因。
2. out.[PFC].png, out.[PFC].pdf, out.[PFC].xls GO富集气泡图,富集条形图,有向无环图,只展示富集的GO term(即p-value小于0.05),如果没有小于0.05的结果则表示无该文件。可以在xls结果中查看,结果对应网页结果,包含GOid,GO功能描述,基因占比,背景基因占比,P值,Q值,以及对应的基因id。
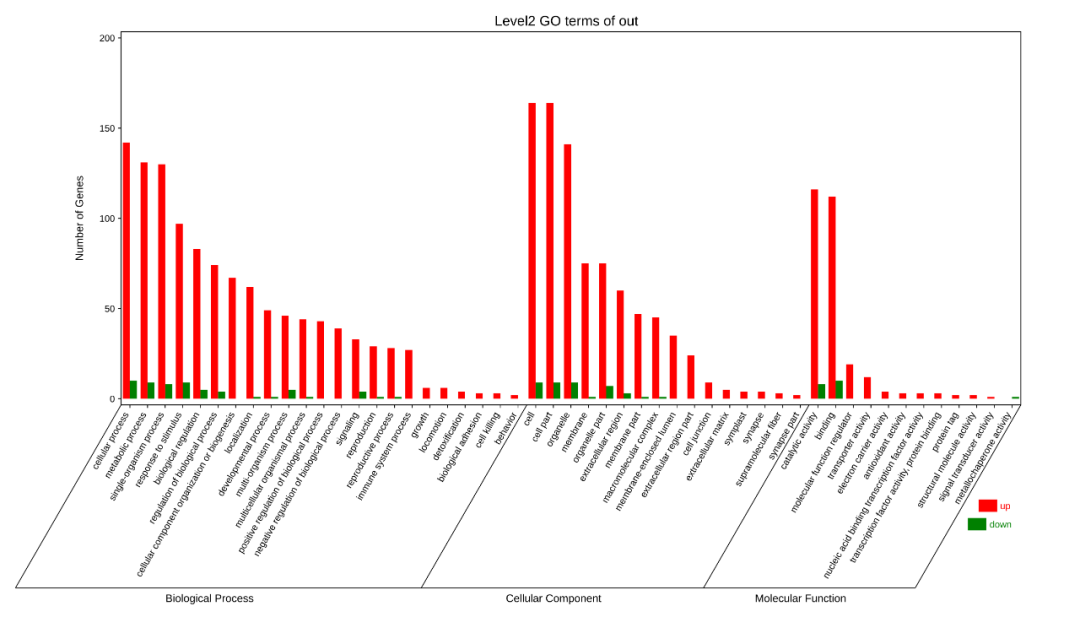
3、out.secLevel.svg/png的图片结果如下图所示,是GO二级分类的统计图,统计了GO二级分类中各个类别中用于富集的基因数量。统计结果在xls表格中。表格内容包括Ontology、Class(GO二级分类)、基因数量、具体基因id。
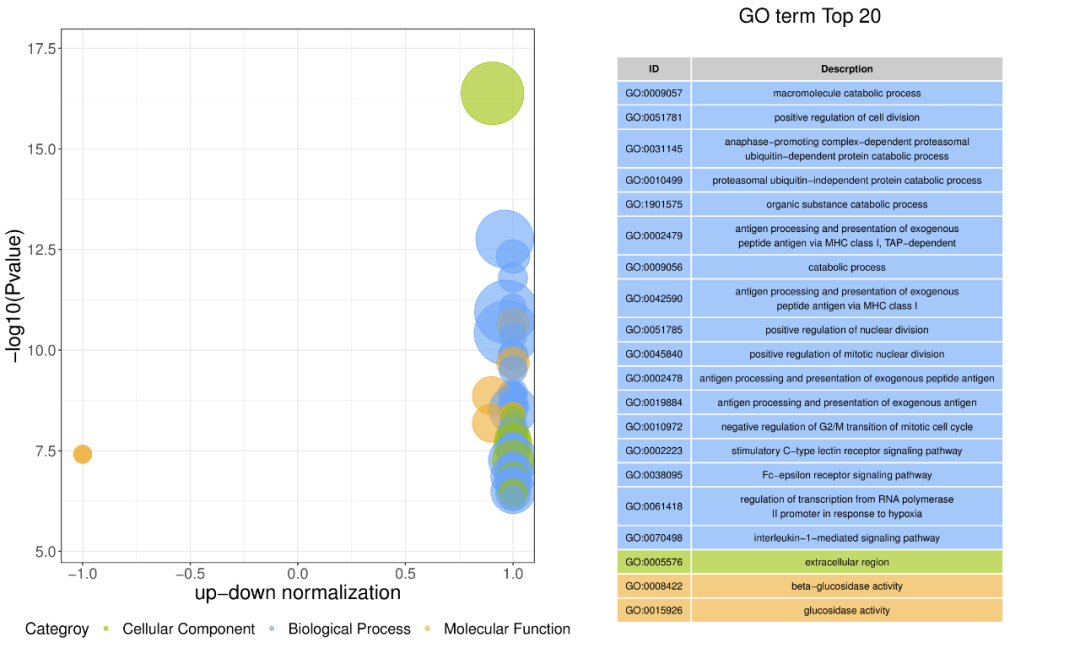
4. out.bubble/bubble_sp.png/pdf的图像结果如下图所示生物信息学在线软件,为GO富集分析的z-score气泡图。纵轴为-log10(Pvalue),横轴为上下标准化值(差异上调基因数与差异下调基因数之差占总差异表达基因数的比例);黄线代表Q/Pvalue=0.05的阈值;右侧为Q/P值前20的terms/pathways列表,不同颜色代表不同的Ontology/A类。
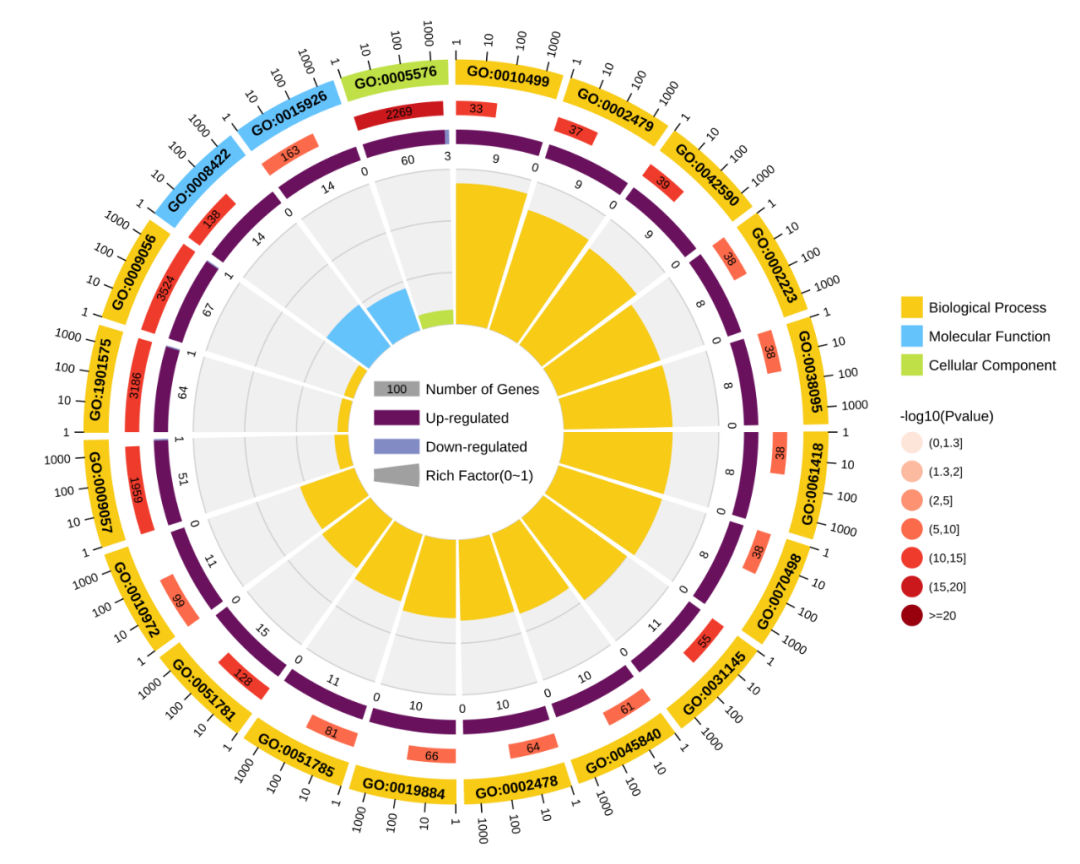
5、out.circular.png/svg的图片结果如下图所示,是GO富集分析的圆形图,从外到内共有四个圆圈:
第一个圆圈:富集类别,外圈为基因数量的坐标刻度,不同颜色代表不同的类别;
第二个圆圈:该类在背景基因中的数量及Q值或P值,基因越多,条形越长,值越小,颜色越红;
第三个圆圈:上调和下调基因比例的柱状图。深紫色代表上调基因比例,浅紫色代表下调基因比例。具体数值在下面展示。当输入的差异调控基因数只有一列(不区分上调和下调基因)时,第三个圆圈展示的是前景基因总数。
第四圈:每个类别的RichFactor值(该类别中前景基因个数除以背景基因个数),背景辅助线每个小格代表0.1。
KEGG富集分析工具
KEGG 整合了基因组、化学和系统功能信息,将从完全测序的基因组中获得的基因目录与细胞、物种和生态系统层面的更高级别的系统功能联系起来。
在KEGG主界面的搜索框中输入Thyroid hormone,点击Go,会出现三张图谱,点击map04919,即可获取数据库中关于甲状腺激素信号转导(PATHWAY: map04919)的相关信息。
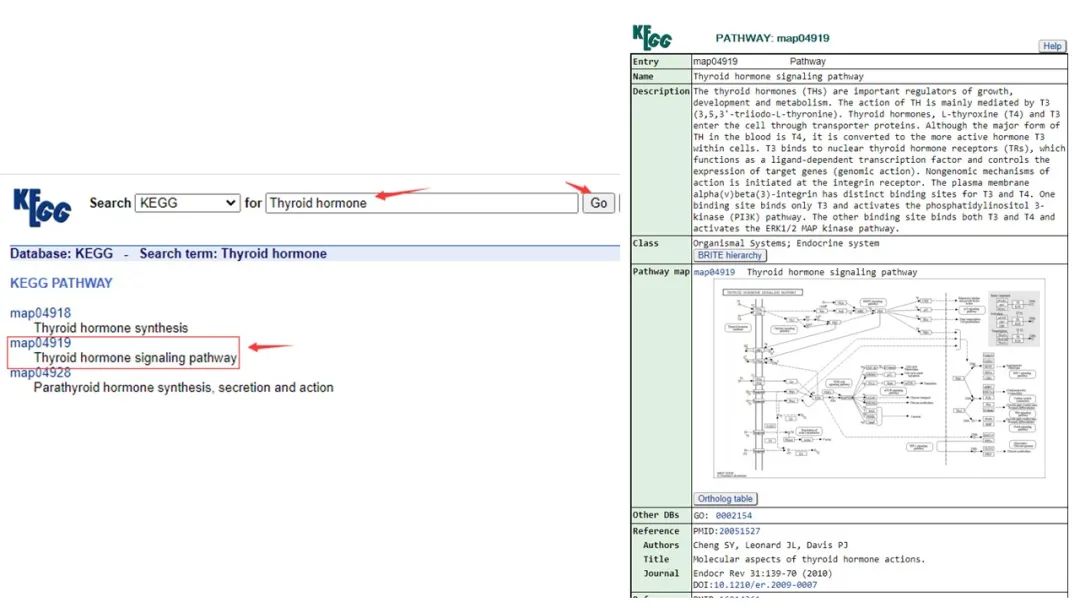
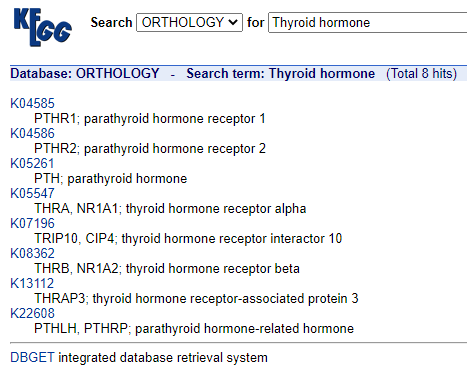
如何查看参与该代谢途径的所有基因?Ortholog列显示参与该代谢途径的所有已知基因。
我们感兴趣的基因参与了这条代谢途径,那怎么用图来表达出来呢?这就需要用到KEGG里的mapping工具了。返回KEGG主界面,在下面的分析与检索工具中选择KEGG Mapper,然后选择Search Pathway,这个就是我们要使用的mapping工具。在select栏中选择reference,在输入框中输入对应基因的ko编号(注意不是基因ID),ko编号后面写上要显示的颜色(默认是粉色),最后点击下面的Exec按钮就可以得到最终的结果了。最后点击结果最下方的map01200,最终就可以生成我们需要的代谢途径图了。

基因组学
基因组杂合性评估
GenomeScope 可以处理一些高度复杂的基因组,比如高杂合度(菠萝,>1%),或者多倍体(8倍体甘蔗),或者非常大的基因组(小麦是16G)。它的作用是通过分析k-mer计数的分布来提供基因组的一些基本信息:基因组大小、基因组杂合度、基因组重复序列比例。
无需安装 GenomeScope,只需安装 jellyfish。
#发布
##Count kmers using jellyfish$ jellyfish count -C -m 21 -s 1000000000 -t 10 *.fastq -o reads.jf##Export the kmer count histogram$ jellyfish histo -t 10 reads.jf > reads.histo##Upload reads.histo to GenomeScope
阿格里戈 v2.0
专注于农业领域的在线分析工具及数据库,已更新至2.0版本,支持45个物种、292种数据类型,拥有基因富集分析、基因富集参数分析、基因富集分析交叉比对等多种工具,可以方便、快速地获得基因富集分析结果。
下面就此网站的使用方法作一简单介绍。

以最常见的基因富集分析为例,点击Species,选择物种(这里以拟南芥为例),点击[ANALYSIS TOOL]选择Singular Enrichment Analysis (SEA),在列表框中输入差异表达基因的ID,点击Submit即可得到结果。如果物种列表中没有对应的物种,可以选择自定义模式,但必须同时输入基因ID和GO登记号。
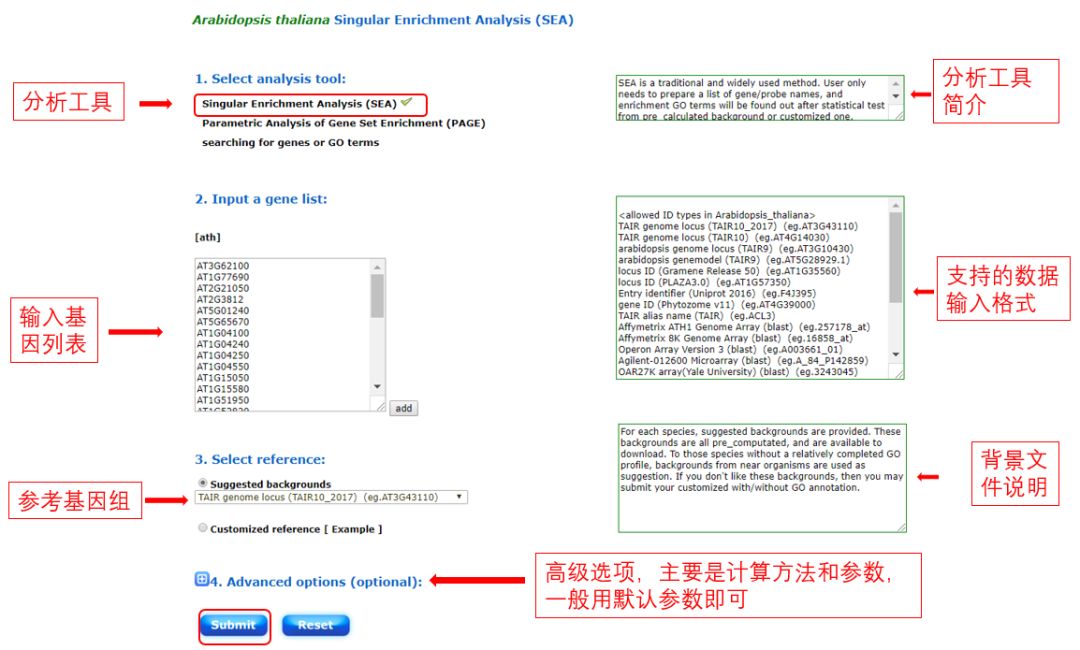
结果文件包含四个部分:

1.分析结果摘要包括物种、参考基因组、显著富集的GO通路数量等信息。
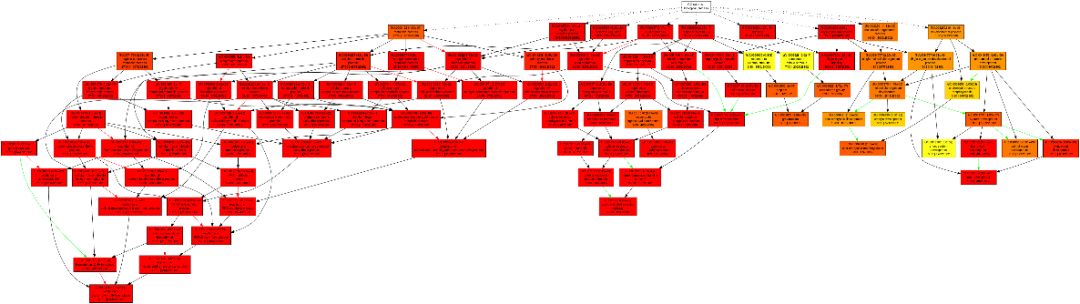
2. 层次图。整个图片分为三个GO类别,分别是生化工程、分子功能和细胞内成分。用户可以提取自己需要的部分,选择图片参数后点击【生成】即可生成图片。每个框代表一条GO通路,并有相应的描述。颜色越深,富集程度越高;
3.条形图展示的是显著富集的GO通路,设置好参数后,点击【generate】生成图片,每条条形图代表一条显著富集的代谢通路。

4. 显著富集的GO通路的详细信息生物信息学在线软件,包括p值,GO通路描述等。
