本文目录导航:
GWAS-1 简介-翻译
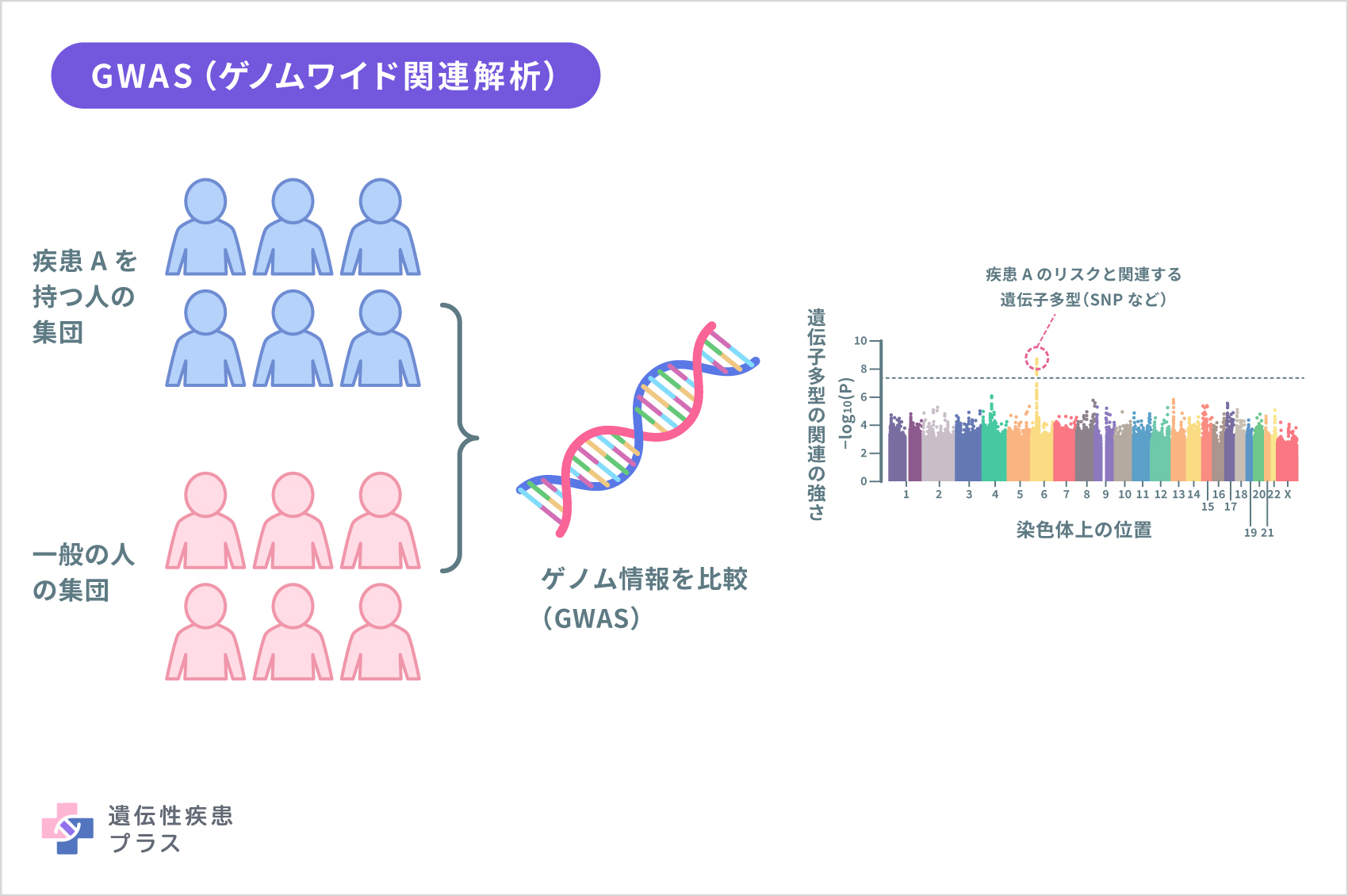
GWAS(genome-wide association study)关键用于钻研相关性状的关键效应。
其思维是应用笼罩全基因组的高度密度SNP标志,经过对每个SNP标志或SNP单倍型与性状的关联剖析,间接找到影响性状的QTN或与数量性状核苷酸(quantitative trait nucleotide,QTN)处于高度连锁不平衡的SNP。
经过必定算法关联SNP与性状的相关,这须要对结果启动度量,其有多种算法。
关于SNP的从编码也有多种,关键的是加性编码,其中SNP的基因型用0、1或2示意,以批示非参考等位基因的数量。
其余或者的编码是显性编码,其中纯合指代基因型编码为0,其余基因型编码为1,而隐性编码,其中纯合代替基因型编码为1,其余基因型编码为0 1.1 数量性状理论经常使用狭义线性模型(GLM)方法启动剖析,最经常出现的是方差剖析(ANOVA),它相似于带有分类预测变量的线性回归,在这种状况下是基因型类别。
经常使用单个SNP启动ANOVA的假定是,任何基因型组的特色均值之间没有差异。
GLM和ANOVA的假定是:1)性状是正态散布的;2)每组内的特色差异相反(各组是同方差的);3)组是独立的。
1.2 理论经常使用列联表法或逻辑回归剖析二分病例/对照特色。
列联表测验审核并测量在表型和基因型类别之间没有关联的零假定下预期的独立性偏向。
该测试最普遍的方式是盛行的卡方测验(以及相关的费舍尔准确测验)。
Logistic回归是线性回归的扩展,其中线性模型的结果经常使用逻辑函数启动转换,该逻辑函数可依据基因型类别预测案例形态的或者性。
Logistic回归理论是首选方法,由于它准许对临床协变量(和其余要素)启动调整,并且可以提供调整后的比值比来。
NP的基因型也可以分为基因型类别或模型,例如显性,隐性,乘性或加性模型。
编码的不同,会给钻研带来不同的结果。
协变量调整可缩小由于钻研伪影或钻研设计中的偏向而形成的虚伪关联,但是调整是以经常使用或者影响统计效用的额外自在度为代价的。
当用作协变量时,这些得分会针对数据中的庞大后人效应启动调整。
关于不同遗传背景的集体混合在一同是影响WAS剖析结果牢靠性的关键要素之一。
普通对一切数据启动PCA剖析,检查前两个PCs的图。
假设分层,须要校对,目前关键的方法有:基因组管理法(genomic control, GC), 结构关联法(structured assocaiation, SA)和主成分剖析法(principal componets). Q-Q图: 以每个SNP的测验统计量的观察值和在原假定(SNP与性状有关)下的希冀值为数据对。
P 值的曼哈顿图都是以-lg(P)示意Y值。
关于每个统计测验,都会生成一个p值,即看到一个测验统计量等于或大于观察到的测验统计量(假设原假定为真)的概率。
这实践上象征着较低的p值示意假设没有关联,则看到此结果的时机十分小。
多重测验可造成I型失误扩展和假阳性关联,因此须要对多重检测校对。
那如何对多重测验启动校对是GWA钻研所面临的关键疑问之一。
目前罕用的方法有:Bonferroni校对,递减调整法(step-down adjustment),数据重排法(data permutaton),Benjamini-Hochberg, 和管理失误发现率法(false discovery rate, FDR)等. Bonferroni校对是最激进和严厉的一种,但是运行较为普遍,公式: Ps = γ / N, Ps是每次检测欲到达的清楚要求的P值的阈值,γ 是所要求的总的范I性失误的概率, N是实践剖析中经常使用的SNP数 统计测验理论被称为有效测验,假设p值低于预约义的alpha值(简直一直设置为0.05),则有效假定会被拒绝。
这象征着在5%的状况下,原假定实践上是真的,而咱们检测到假阳性,则原假定被拒绝。
该概率是相关于单个统计测验而言的;就GWAS而言,启动了数十万至数百万次测试,每个测试都有其自己的假阳性概率。
因此,在整个GWAS剖析中发现一个或多个误报的累积或者性要高得多 批改多重测试的最简双方法之一是Bonferroni批改。
Bonferroni校对将alpha值从α= 0.05调整为α=(0.05 / k),其中k是启动的统计测验的次数。
关于经常使用500,000个SNP的典型GWAS,SNP关联的统计显着性应设置为1e-7。
此校对是最激进的,由于它假定500,000的每个关联测试均独立于一切其余测试-由于GWAS标志之间的连锁不平衡,这一假定理论是不正确的。
调整误报率(alpha)的另一种方法是确定误发现率(FDR)。
失误发现率是对关键结果(理论为alpha = 0.05)中误报所占比例的预计。
在GWAS数据集中没有真实关联的零假定下,关联测试的p值将遵照平均散布(从0到1平均散布)。
FDR程序最后由Benjamini和Hochberg开发,从实质上 纠正 了预期的失误发现数量,从而提供了对那些被称为“严重发现”的真实结果的预计[[33]]。
这些技术已宽泛运行于GWAS,并以多种方式扩展[[34]]。
置换测试是在GWAS中建设关键性的另一种方法。
虽然计算量大,但置换测试是在原假定为真时为给定数据集生成测试统计量的阅历散布的间接方法。
这是经过将每个集体的表型随机从新调配给数据集中的另一个集体来成功的,从而有效地冲破了数据集的基因型与表型之间的相关。
数据的每次随机重调配代表在原假定下对集体的一个或者采样,并且此环节重复了预约的次数N以生成分辨率为N的阅历散布,因此N为1000的置换环节给出了阅历p 1/1000内-VALUE 日 小数点后一位。
曾经开发了几种软件包来口头GWAS钻研的置换测试,包括盛行的Plink软件[[35]],PRESTO [[36]]和PERMORY [[37]] 全基因组关联钻研为审核整个基因组的遗传变异之间的相互作用提供了庞大的时机。
但是,多场合剖析并不像启动单场合测试那样便捷,并且提出了许多计算,统计和后勤方面的挑。
由于大少数GWAS基因型介于500,000个和一百万个SNP之间,所以即使关于高效算法,审核SNP的一切成对组合也是一种计算上辣手的方法。
处置此疑问的一种方法是缩小或过滤基因型SNP的汇合,从而消弭冗余消息。
过滤SNP的一种便捷而通用的方法是,依据恣意关键性阈值从单SNP剖析当选用一组结果,并详尽地评价该子集中的相互作用。
但是,这或者很风险,由于基于关键效应选用要剖析的SNP将阻止检测到某些多位点模型-所谓的“纯上位”模型,其边沿效应在统计上是无法检测的。
经常使用这些模型,遗传力的很大一局部都集中在交互而不是关键成果上。
换句话说,标志物的特定组合(仅标志物的组合)惹起疾病风险的显着变动。
这种剖析的好处在于,它对所选SNP汇合内的相互作用启动了无偏性剖析。
与剖析一切或者的标志组合相比,它在计算和统计上也更容易处置。
另一种战略是将SNP组合的审核限度为属于已建设的动物学环境(例如生化路径或蛋白质家族)中的那些组合。
由于这些技术依赖于结构化动物医学常识的电子存储库,因此它们理论将生成SNP-SNP组合的动物消息引擎与评价GWAS数据集中组合的统计方法结合经常使用。
例如,动物过滤器方法经常使用了各种公共数据源,以及逻辑回归和多要素降维方法[[40]], [41] 。
雷同,INTERSNP经常使用逻辑回归,对数线性和列联表方法评价SNP-SNP相互作用模型 [42] 。
简而言之,复制钻研的普通战略是尽或者重复启动GWAS确实定和设计,但仅审核在GWAS中发现的显着遗传效应。
两项钻研中分歧的效应可以标志为重复效应。
荟萃剖析的基本原理是,一切归入的钻研都测验了相反的假定。
因此,每个归入钻研的总体设计应相似,并且钻研水平的SNP剖析应在一切钻研中遵照简直相反的程序(参见Zeggini和Ioannidis[47] 以取得杰出的评价)。
确定每个站点蕴含哪些SNP的品质管理程序以及一切协变量调整均应规范化,并且多个站点之间临床协变量和表型的测量应坚持分歧。
一切钻研的样本集都应该是独立的–由于钻研人员经常将相反的样本奉献给多个钻研,因此应该经常审核这一假定。
雷同,一个极端关键且有点费事的后勤疑问是要确保一切钻研报告相关于经常出现基因组构建和参考等位基因的结果。
假设一项钻研报告了其相关于等位基因的结果,此SNP的荟萃剖析结果或者并不关键,由于两项钻研的成果相互对消。
思考到一切这些要素,很少能找到在一切条件下都齐全婚配的多项钻研。
因此,经常在荟萃剖析中对钻研异质性启动统计量化,以确定钻研之间的差异水平。
钻研异质性最盛行的度量是I2指数I2指数在最近的钻研中更受青眼。
由荟萃剖析得出的系数具备可变性(或误差),并且I指数示意该可变性的近似比例,这可以归因于钻研之间的异质性。
I2值分为低(<25),中(> 25和<75)和高(> 75)异质性,并且已被提议作为一种识别或者应该从荟萃剖析中删除的钻研的方法。
关键的是要留意,这些统计数据应被用作识别或者与荟萃剖析中其余假定不同的基础假定的钻研的指点,就像离群剖析被用来识别影响力过大的观念一样。
但是,与意外值一样,仅在有清楚理由基于钻研参数启动钻研时才应将其扫除在外–不只仅是由于统计数据标明该钻研会参与异质性。
否则,旨在缩小荟萃剖析异质性的无法知统计程序将参与失误的发现 google翻译文章:Bush WS, Moore JH. Chapter 11: Genome-wide association studies. PLoS Comput Biol. 2012;8(12):e. doi: 10.1371/.. Epub 2012 Dec 27. PMID: ; PMCID: PMC. “Exploration of a diversity of computational and statistical measures of association for genome-wide genetic studies”()
GWAS系列 | 带你读懂全基因组关联钻研(一)
全基因组关联钻研(GWAS)是一种识别遗传区域与特定性状或疾病关联的方法。
经过检测基因组数百至上千万遗传变异,GWAS可找出与特定表型或疾病清楚关联的变异位点。
该方法已被用于鉴定各类表型和疾病关联,相关遗传变异数量随样本量参与而增长。
GWAS运行宽泛,包括深化了解表型的动物学机制、遗传力预计、计算遗传相关性、临床风险预测、药物开发以及推断泄露要素与终局间的因果咨询。
GWAS试验流程理论包括搜集受试者基因组消息、基因分型、品质管理与数据荡涤、关联统计测验、GWAS荟萃剖析以及独立验证。
品质管理确保数据准确,而荟萃剖析整合多个钻研结果,增强关联证据。
独立验证队列的经常使用确保发现结果的牢靠性。
在GWAS钻研中,可应用的地下数据库丰盛,如UK Biobank、HapMap和1000 Genomes等,提供了少量基因型和表型数据,支持钻研者启动深化剖析。
舒桐科技专一于GWAS钻研,提供翻新技术、一站式基因编辑和分子诊断平台,以及CGT原料规模化消费才干,旨在提升GWAS产品,为科研与动物医药企业提供高效服务。
如需进一步探讨,请咨询咱们。
GWAS基本剖析内容
之前给大家大抵引见了GWAS在临床生信剖析中的详情,包括一些基本概念,原理和留意事项(出门左手边—> 临床动物消息学中的GWAS剖析 ),这次详细讲讲 GWAS基本剖析内容及结果解读 (这篇也是我在百迈客云课堂的团体学习笔记,有兴味学习的可以去购置课程 百迈客GWAS动物消息培训课程 )另外参考文献及配图来自 GWAS基本剖析内容 1)按分型百分比过滤 普通剔除缺失率在20%以上的位点,假设数据量比拟大可以放宽到50%2)按等位基因频率过滤 去除第二等位基因频率小于5%的位点,假设数据量比拟大可以放宽到1%3)多等位位点的过滤 依据软件的须要,有些软件不支持多等位位点4)哈迪温伯格平衡过滤 人类case/control中普通将不合乎哈迪温伯格平衡的位点过滤掉,动植物不经常使用该过滤5)极端表型的去除 最低饱和标志量=基因组大小/LD衰减距离 密度越大越好:检测到配置位点的概率增大;处于同一block的位点相互验证 可以依据LD衰减距离来选择候选基因高低游的范畴 1)目标:对集体结构和亲缘相关启动评价以确定经常使用的统计模型和取得相应的矩阵2)评价内容(遗传上差异过大应剔除,相似性高的保管其一) 集体结构: 构建系统发育树 (必备)同一物种内序列差异不大构建NJ树(mega),序列差异较大,不同种构建ML树(RAxML),贝叶斯树(ExaBayes) model-base的集体结构剖析 主成分剖析 亲缘相关:相似性系数热图、遗传距离 3)集体结构和亲缘相关是造成关结合果产生 假阳性 的两个关键要素(课程里谢坤大牛用实例说明了集体结构和亲缘相关的选用对结果的庞大影响,比如玉米Dwarf8基因 Lessons from Dwarf8 on the Strengths and Weaknesses of Structured Association Mapping )1)选用正确的统计方法: 小标志量:便捷的t-test或ANOVA case/control品质性状:卡方测验, OR测验,逻辑回归 数量性状:普通经常使用多种模型(GLM/MLM/EMMAX/fast-LMM)同时剖析2)确定清楚性阈值 Bonferroni correction 3)结果解读 可视化:曼哈顿图分位点图(随机预测出的P和实践算进去P的比拟,能否意外清楚)4)若结果不清楚: 或者要素及处置方法 : 性状调查不准确——提供准确表型 性状受环境影响大——多年多点重复 性状由多个小效应位点管理——参与样本量 模型检测效能power不够——改换适合的模型 标志密度不够——愈加标志量 真实不行就疏忽阈值,选用有清楚的峰值区域启动验证 取得清楚位点后: 1)启动LD block剖析,确定候选区间的范畴 2)对候选区间内的基因做配置注释(nr,GO,KEGG等) 3)清楚位点能否位于编码区,能否是非同义突变 4)同源剖析,结合其余物种对应的同源基因的配置来猜想候选基因的配置 RT-PCR、蛋白表白、转基因配置验证、RNA搅扰 高分文章实例: OsSPL13 controls grain size in cultivated rice
